Chemprop¶
Molecular Property Prediction¶
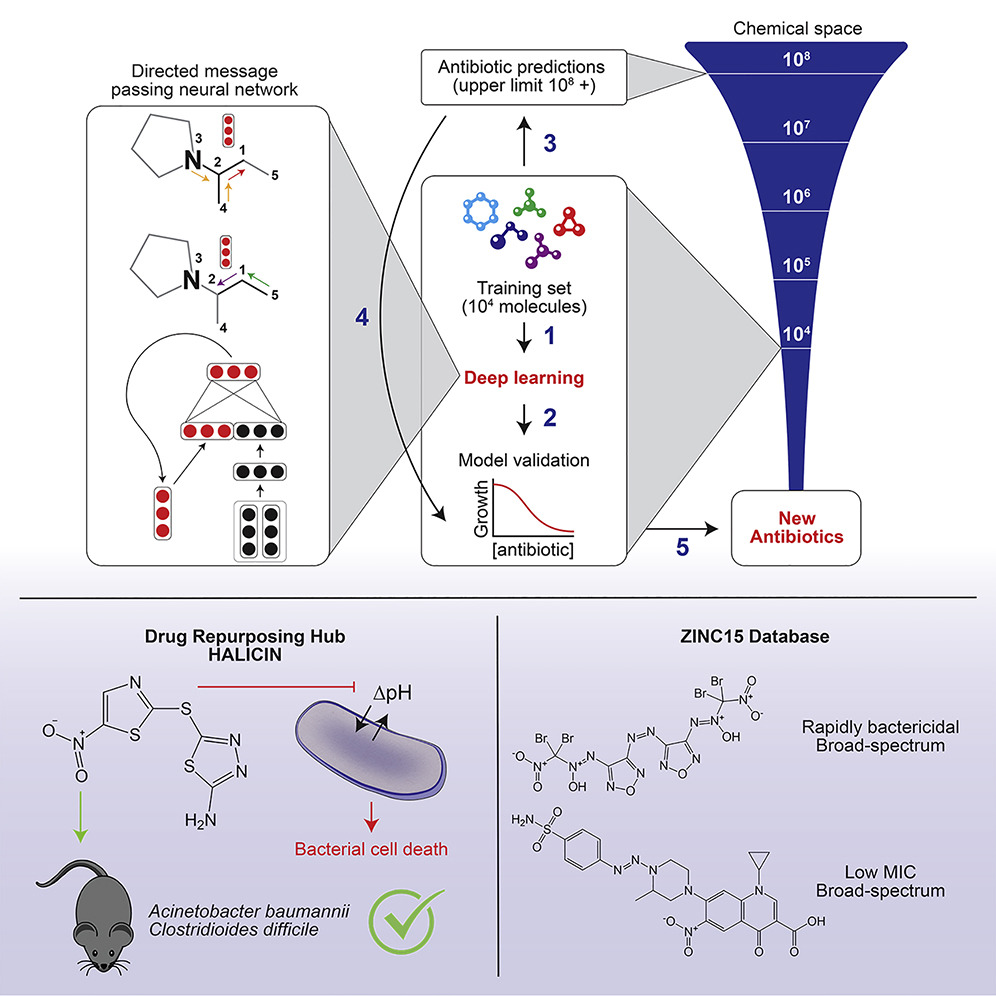
neural networks for molecular property prediction as described in the paper Analyzing Learned Molecular Representations for Property Prediction and as used in the paper A Deep Learning Approach to Antibiotic Discovery for molecules and Machine Learning of Reaction Properties via Learned Representations of the Condensed Graph of Reaction for reactions.
Analyzing Learned Molecular Representations for Property Prediction¶
Takeaways¶
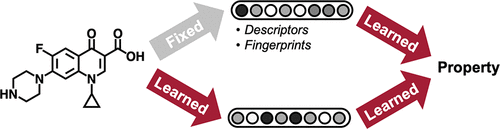
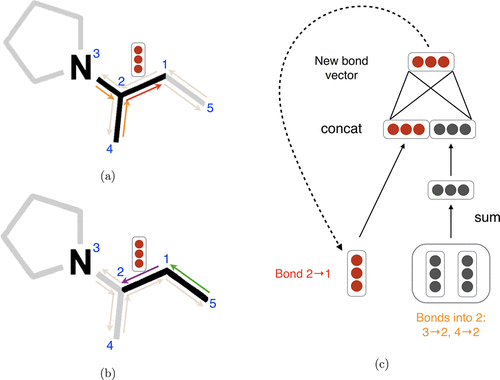
The model is characterized by two unique features: (1) It employs a hybrid representation that integrates both convolutions and descriptors. This structure enables the model to learn task-specific encodings with flexibility, while also benefiting from the robust prior provided by fixed descriptors. (2) The model learns to build molecular encodings through the use of convolutions centered on bonds rather than atoms, which eliminates the need for redundant loops during the message-passing stage of the algorithm.
Additionally, we demonstrate that using a scaffold-based division of training and testing data is an effective approximation of the industry-standard temporal split, as measured by relevant metrics.
The selection of hyperparameters is critical to the performance of the model. Depending on the choice, performance improvements can range from a modest 2-5% to a substantial 37%.
Across the seven public datasets, there is no single baseline model that consistently outperforms the others.
The direction in which messages are passed is significant. The model uses one-hot encodings for both atom and bond features.
Hyperparameter tuning is performed using Bayesian Optimization through the Hyperopt Python package.
Incorporating additional features results in varying performance across different datasets. Some datasets exhibit significant improvements, while others show a decline in performance. This could be attributed to the additional features either enhancing the model’s understanding or causing confusion and distraction in certain tasks.
One limitation of the model is that it does not make use of 3D structural information.
A Deep Learning Approach to Antibiotic Discovery¶
Using chemprop model.
Features¶
Atom features: atomic number, number of bonds for each atom, formal charge, chirality, number of bonded hydrogens, hybridization, aromaticity, atomic mass.
Bond features: bond type (single/double/triple/aromatic), conjugation, ring membership, stereochemistry.
Additional molecule-level features¶
200 additional molecule-level features computed with RDKit.
Ensembling¶
ensemble of 20 models, with each model trained on a different random split of the data.
four phases
(1a) a training phase to evaluate the optimized but non-ensembled model
(1b) training the ensemble of optimized models
a prediction phase
a retraining phase
a final prediction phase.
Tanimoto similarity: quantify the chemical relationship between molecules
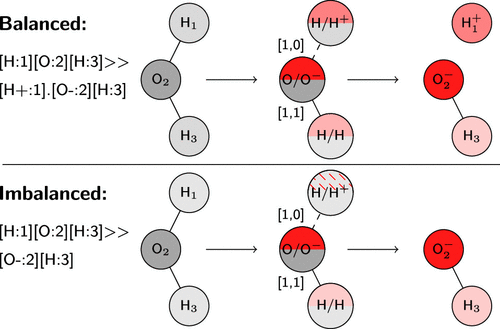
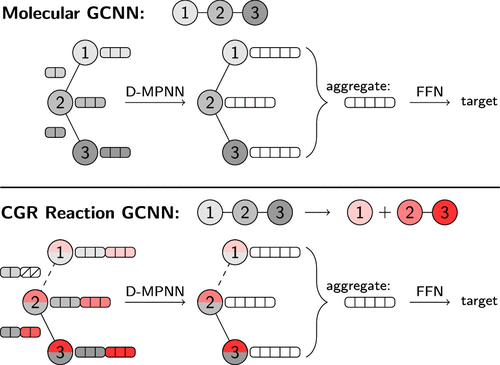
Machine Learning of Reaction Properties via Learned Representations of the Condensed Graph of Reaction¶
In the case of D-MPNNs, messages are associated with directed edges instead of vertices, in contrast to regular MPN architectur
atom features : one-hot encoding of the atomic number, degree, formal charge, chirality, number of hydrogens, hybridization, and aromaticity o the atom, as well as the scaled atomic mass, resulting in vectors f length 133. bond features : the bond type, whether the bond is conjugated, in a ring, and contains stereochemical information, resulting i vectors of leng.
The reliability of the imputed data can be learned by comparing the features with the structure of th graphth 283es